Step-by-step Illustration¶
In the Quick Start Example section, we have shown the simplest all-in-one command line of
performing the pipeline with a wrapper script imargi_wrapper.sh. Here we go inside of the quick example to illustrate
the details of iMARGI Pipeline. If you want to customize your pipeline, you can run each step separately with
corresponding tools.
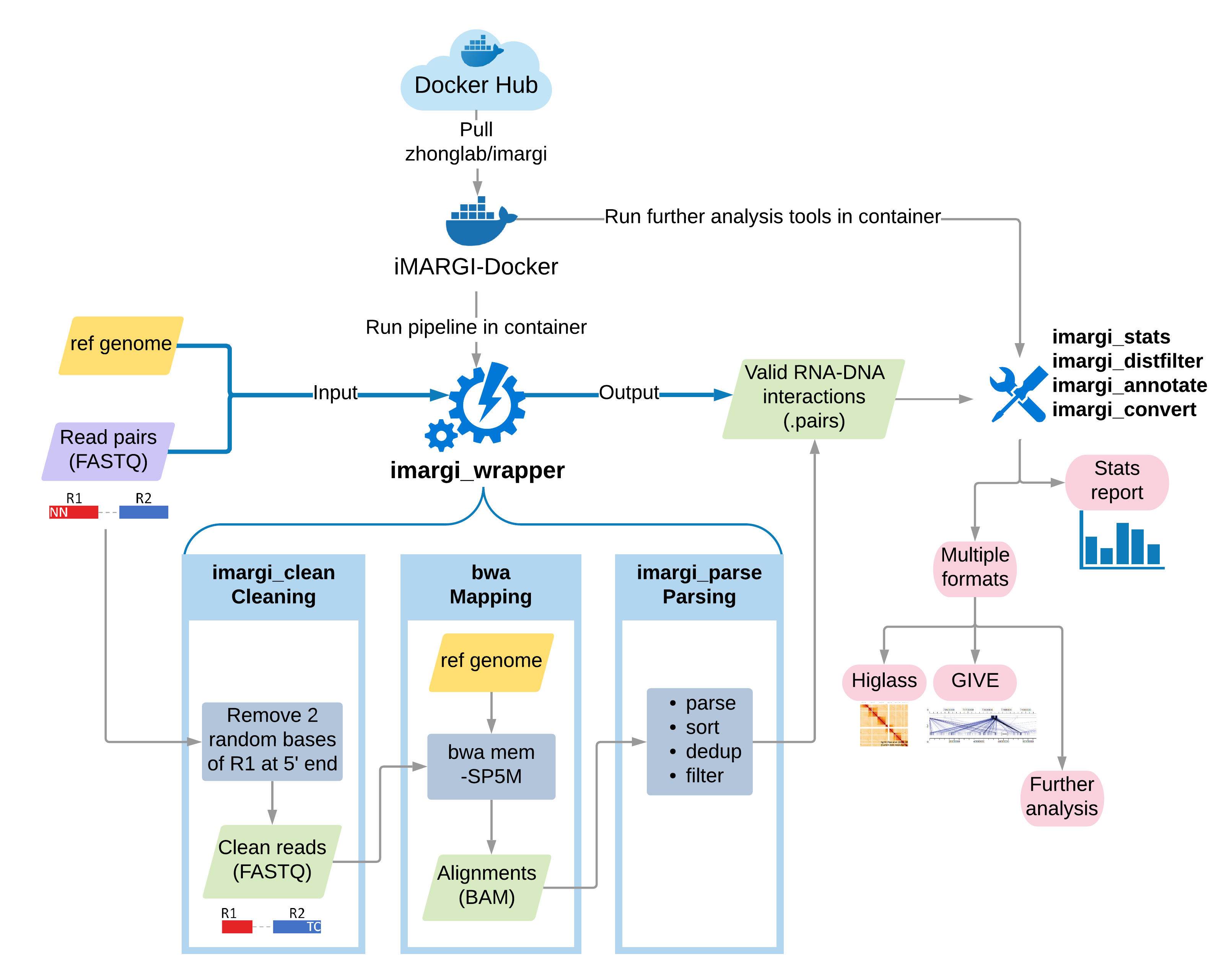
- Step-by-step Illustration
The imargi_wrapper.sh Running Command¶
In the quick example section, we use the following command of imargi_wrapper.sh, whose input
are only sequencing read pairs FASTQ files and reference genome sequence FASTA file.
docker run --rm -t -u 1043 -v ~/imargi_example:/imargi zhonglab/imargi \
imargi_wrapper.sh \
-r hg38 \
-N test_sample \
-t 4 \
-g ./ref/GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta \
-i ./ref/bwa_index/bwa_index_hg38 \
-1 ./data/sample_R1.fastq.gz \
-2 ./data/sample_R2.fastq.gz \
-o ./output
We only set a few arguments in the command which are described below, and use default settings for other arguments.
-r: Name of reference genome. It will be used in header of .pairs format file and names of new generated reference files.-N: Name of sample. It will be used in names of final output files.-t: Maximum CPU cores.-g: Reference genome sequence FASTA file.-i: BWA index files.-1and-2: Sequencing read pairs FASTQ files.-o: Output directory
In fact, there are more arguments of imargi_wrapper.sh can be set, you can check the full arguments in the
Command-line API section. Here we want to remind you the following three arguments of required
reference files, including chromosome sizes file, bwa index and restriction fragments bed format file. Because we didn’t
set these arguments in the command line, imargi_wrapper.sh automatically generated them. If you have already got these
files, you can use these arguments and it will save you some time and disk space. When the three arguments are all set
correctly, the -g argument can be ignored.
-i: BWA index files.-c: Chromosome sizes file-R: Restriction fragments bed format file
The following sections are detail illustrations of how imargi_wrapper.sh works inside of the iMARGI Docker
container.
Generating Required Reference Files¶
As we only input reference genome sequence FASTA file with -g argument, so imargi_wrapper.sh will automatically
generate other required reference files, such as chromosome sizes, bwa index and restriction fragments.
Generating Chromosome Sizes File¶
| Type | Description/Tool | Files/Key Parameters |
|---|---|---|
| Tool | samtools faidx |
<ref_genome_FASTA> |
| Input | ref genome sequence in FASTA | ./ref/GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta |
| Output | chromosome sizes file | ./ref/chrom.sizes.hg38.txt |
The imargi_wrapper.sh uses samtools faidx to generate the chromosome sizes file from reference genome sequence FASTA
file. The command lines used in imargi_wrapper.sh is:
cd ~/imargi_example/ref
samtools faidx GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta
cat GRCh38_no_alt_analysis_set_GCA_000001405.15.fa.fai |awk 'BEGIN{OFS="\t"}{print $1,$2}' >chrom.sizes.hg38.txt
Building BWA Index¶
| Type | Description/Tool | Files/Key Parameters |
|---|---|---|
| Tool | bwa index |
-p <idxbase> <ref_genome_FASTA> |
| Input | ref genome sequence in FASTA | ./ref/GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta |
| Output | a folder of bwa index files | ./ref/bwa_index |
BWA is used to map sequencing reads to reference genome, so we need to construct bwa index for reference genome first.
Here is the command lines used in imargi_wrapper.sh:
cd ~/imargi_example/refmkdir
bwa index -p ./ref/bwa_index/bwa_index_hg38 ./ref/GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta
Generating Restriction Fragment File¶
| Type | Description/Tool | Files/Key Parameters |
|---|---|---|
| Tool | imargi_rsfrags.sh |
- r <ref_genome_FASTA> -c <chromSize_file> -e <enzyme_name> -C <cut_position> |
| Input | ref genome sequence in FASTA | ./ref/GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta |
| Output | restriction fragment BED file | ./ref/AluI_frags.bed.gz |
AluI enzyme is used to digest genome in iMARGI, whose cutting site is AG^CT. The restriction fragments are used for
filtering out read pairs sequenced from non-RNA-DNA-ligation fragments. The imargi_wrapper.sh uses imargi_rsfrags.sh
tool to generate the restriction fragment bed (gzip compressed) format file.
imargi_rsfrags.sh \
-r ./ref/GRCh38_no_alt_analysis_set_GCA_000001405.15.fasta \
-c ./ref/hg38.chrom.sizes \
-e AluI \
-C 2 \
-o ./ref/iMARGI_AluI_rsfrags.bed.gz
Cleaning: clean read pairs¶
| Type | Description/Tool | Files/Key Parameters |
|---|---|---|
| Tool | imargi_clean.sh |
-1 <fastq.gz_R1> -2 <fastq.gz_R2> -o <output_dir> -t <threads> |
| Input | raw paired FASTQ files | ./data/sample_R1.fastq.gz ./data/sample_R2.fastq.gz |
| Output | clean paired FASTQ files | ./output/clean_fastq/clean_test_sample_R1.fastq.gz ./output/clean_fastq/clean_test_sample_R2.fastq.gz |
According to the design of iMARGI sequencing library construction, there are two random nucleotides NN at the 5’ end
of R1 read. It will affect mapping, so we remove those two random bases before mapping.
As the 5’ end of R2 is DNA restriction enzyme digestion site, which is AluI in iMARGI experiment (AG^CT), so the DNA
end should always start with “CT”. Hence, we can use this to filter out non-ligated RNA contaminations.The
imargi_clean.sh tool has an option -f <filter_CT>, which can be used to filter read pairs based on the 5’
end sequence of R2 reads as -f CT. The advantage of of such hard filtering is reduce the computational cost.
But the disadvantage is that it’s not flexible and causes false negative. So we don’t use the filter, and the filter
based on restriction fragments is applied in parsing step within the imargi_parse.sh tool.
The command of cleaning used in the imargi_wrapper.sh is:
bash imargi_clean.sh \
-1 ./data/sample_R1.fastq.gz \
-2 ./data/sample_R2.fastq.gz \
-o ./output/clean_fastq/ \
-t 16
Mapping: map reads to genome¶
| Type | Description/Tool | Files/Key Parameters |
|---|---|---|
| Tool | bwa mem |
-SP5M -t <threads> <idxbase> <in1.fq> <in2.fq> |
| Input | clean paired FASTQ files | ./output/clean_fastq/clean_test_sample_R1.fastq.gz ./output/clean_fastq/clean_test_sample_R2.fastq.gz |
| Output | BAM file | ./output/bwa_output/test_sample.bam |
We use bwa mem with -SP5M parameters to map cleaned read pairs to reference genome. The -SP5M options are very
important, which allows split alignments. The output BAM file name is based on the -N argument of imargi_wrapper.sh,
and the name will be kept in next step.
# bwa mem mapping
bwa mem -t 16 -SP5M ./ref/bwa_index/bwa_index_GRCh38 \
./clean_fastq/clean_test_sample_R1.fastq.gz \
./clean_fastq/clean_test_sample_R2.fastq.gz |\
samtools view -Shb -@ 7 - >./bwa_output/test_sample.bam
Parsing: parse mapped read pairs to valid RNA-DNA interactions¶
| Type | Description/Tool | Files/Key Parameters |
|---|---|---|
| Tool | imargi_parse.sh |
-r <ref_name> -c <chromSize_file> -R <restrict_frags_file> -b <bam_file> -o <output_dir> -t <threads> |
| Input | BAM file | .output/bwa_output/test_sample.bam |
| Output | .pairs file | ./output/final_test_sample.pairs.gz |
The imargi_parse.sh is a wrapper script to parse interaction pairs from BAM file, and do de-duplication and filtering,
then output the final .paris file of valid RNA-DNA interaction map, and by default the intermediate files are also kept
in ./output/parse_temp. There are also some arguments can be used to tweak the thresholds of parsing and filtering.
You can use the default value or customize them.
-Q <min_mapq>: The minimal MAPQ score to consider a read as uniquely mapped. [default: 1]-G <max_inter_align_gap>: Read segments that are not covered by any alignment and longer than the specified value are treated as “null” alignments. [default: 20]-O offset_restriction_site: Offset allowed for restriction fragment filter. [default: 3]-M <max_ligation_size>: Selected sequencing fragment size. [default: 1000]
The command of parsing used in the imargi_wrapper.sh is:
imargi_parse.sh \
-r hg38 \
-c ./ref/hg38.chrom.sizes \
-R ./ref/AluI_frags.bed.gz \
-b ./output/bwa_output/test_sample.bam \
-o ./output \
-t 16 \
-Q 1 \
-G 20 \
-O 3 \
-M 1000 \
-d false \
-D $output_dir/parse_temp
The core tool used in imargi_wrapper.sh is pairtools. Here are some brief descriptions of the usages of pairtools
in our work. If you want to know more, please check the GitHub repo and
documentation of pairtools.
pairtools parse: Parse the types of read pairs based on their mapping results. Output a .pairs file. The parameters--add-columns cigar,--no-flip,--walks-policy 5anyare required for iMARGI data, don’t change it unless you know what you want to do.pairtools dedup: Mark and de-duplications in the.pairsfile.pairtools select: Filter out those read pairs which are marked as duplications, multiple mappings and 5’ most end was not unique mapped. We used a long filter string designed for iMARGI, don’t change it.
There will be pipelineStats_xx.log file in the output directory. It reports some basic statistics of the parsing
results, such as total number of read pairs, unique mapped read pairs and valid RNA-DNA interactions.
Filtering strategies
Besides of filtering out unmapped, duplicated and multiple mapped read pairs, we also use restriction fragments information for filtering out those potential un-ligated RNA or DNA fragment contaminations. We check the 5’ end of mapped read pairs R1 (RNA-end) and R2 (DNA-end), there are three cases based on the relative position of 5’ end and the nearest restriction digestion site, which are denoted as dist1 and dist2, and we have a threshold of offset:
- dist2 > offset: R2 (DNA-end) is not sequenced from a proper restriction fragment, so the sequencing fragment might be un-ligated RNA
- dist2 < offset & dist1 < offset: sequencing fragment might be a un-ligated DNA if the distance of R1 and R2 is less than the maximum sequencing fragment size and the strands of R1 and R2 are opposite
- dist2 < offset & dist1 > offset: proper RNA-DNA ligation
The filtering argument used in pairtools is:
"regex_match(pair_type, \"[UuR][UuR]\") and \ dist2_rsite != \"!\" and \ (abs(int(dist2_rsite)) <= "$offset") and \ (not (chrom1 == chrom2 and \ abs(int(dist1_rsite)) <= "$offset" and \ strand1 != strand2 and \ ((strand1 == \"+\" and strand2 == \"-\" and int(frag1_start) <= int(frag2_start) and \ abs(int(frag2_end) - int(frag1_start)) <= "$max_ligation_size") or \ (strand1 == \"-\" and strand2 == \"+\" and int(frag1_start) >= int(frag2_start) and \ abs(int(frag1_end) - int(frag2_start)) <= "$max_ligation_size"))))"
Although we set several filtering strategies, they still cannot filter out all ‘contaminations’. So we use genomic
distance as a final filter. Filtering out those short-range interactions is better for further analysis. We provide a
tool imargi_distfilter.sh to do genomic distance filter. Please read more in the
further analysis instructions.
Output Files¶
All The Output File List¶
The output files are all in the output directory ~/imargi_example/output.
The final RNA-DNA interaction map is the final_test_sample.pairs.gz file, which is in a compressed .pairs format and
can be used for further analysis. There is a pipeline summary log file, pipelineStats_test_sample.log in the output
directory, which reports the sequence mapping QC result (pass or failed), total processed read pairs number, BWA mapping
stats and number of valid RNA-DNA interaction in the final .pairs.gz file. All other intermediate result files are
also kept in several sub-folders of the output directory.
Besides, if we used the minimum input requirements, the pipeline will automatically generated several required reference files in
the same directory of reference genome instead of the output directory, including chromosome sizes, bwa index and AluI
digestion fragments. When processing new dataset, you can reuse these new generated reference files with corresponding
arguments instead of only using -g argument, which will save you some time and disk space.
The final tree structure of the output directory is shown below.
└── output
├── bwa_output
│ ├── bwa_log_test_sample.txt
│ └── test_sample.bam
├── clean_fastq
│ ├── clean_test_sample_R1.fastq.gz
│ └── clean_test_sample_R2.fastq.gz
├── parse_temp
│ ├── dedup_test_sample.pairs.gz
│ ├── drop_test_sample.pairs.gz
│ ├── duplication_test_sample.pairs.gz
│ ├── sorted_all_test_sample.pairs.gz
│ ├── stats_dedup_test_sample.txt
│ ├── stats_final_test_sample.txt
│ └── unmapped_test_sample.pairs.gz
├── final_test_sample.pairs.gz
└── pipelineStats_test_sample.log
The table below briefly described all the output files.
| Type | Example Items | Directory (relative to output directory) | Format | Description |
|---|---|---|---|---|
| Cleaned FASTQ | clean_test_sample_R1.fastq.gz clean_test_sample_R2.fastq.gz |
./clean_fastq |
paired clean FASTQ | Output of cleaning step, the two random bases at 5' end of R1 were removed |
| Sequencing reads alignments | test_sample.bam |
./bwa_output |
BAM | Output of bwa mapping step |
| BWA mapping log | bwa_log_test_sample.txt |
./bwa_output |
text | BWA mapping log file |
| Intermediate parsing results | sorted_all_test_sample.pairs.gz |
./parse_temp |
.pairs | Sorted all the parsed read pairs |
| Intermediate parsing results | dedup_test_sample.pairs.gz |
./parse_temp |
.pairs | Parsed read pairs after deduplication |
| Intermediate parsing results | duplication_test_sample.pairs.gz |
./parse_temp |
.pairs | Duplicated pairs |
| Intermediate parsing results | unmapped_test_sample.pairs.gz |
./parse_temp |
.pairs | Unmapped pairs (including multiple mapping) |
| Intermediate parsing results | drop_test_sample.pairs.gz |
./parse_temp |
.pairs | Pairs filtered out filtering step |
| Intermediate parsing results | stats_dedup_test_sample.txt stats_final_test_sample.txt |
./parse_temp |
text | pairtools intermediate stats report |
| Final output | final_test_sample.pairs.gz |
./ |
.pairs | Final output of valid RNA-DNA interactions |
| Pipeline stats log | pipelineStats_test_sample.log |
./ |
text | Sequence mapping QC result and stats report |
The iMARGI Pipeline Output .pairs Format¶
The default final output file is final_test_sample.pairs.gz which is in .pairs format. .pairs file format is designed
by 4DN DCIC. You can read its
specification document.
In iMARGI Pipeline, we add some more data columns to the default .pairs format files.
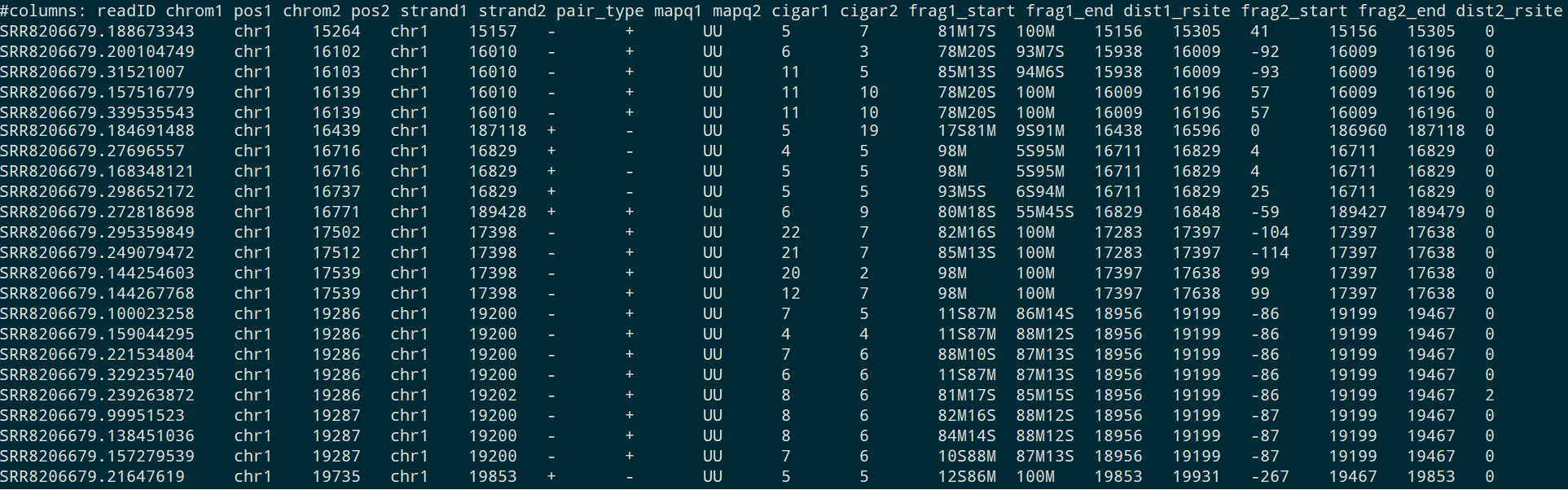
The data column descriptions are listed in the table below. (All the genomic coordinates in .pairs file are 1-based.) The first 7 columns are standard information, 8-18 are extra information. All these column names are reserved.
| column order | name | datatype | description |
|---|---|---|---|
| 1 | readID | string | read ID |
| 2 | chrom1 | string | mapping chromosome of R1 (RNA-end) |
| 3 | pos1 | int | 5' end mapping position of R1 (RNA-end) |
| 4 | chrom2 | string | mapping chromosome of R2 (DNA-end) |
| 5 | pos2 | int | 5' end mapping position of R2 (DNA-end) |
| 6 | strand1 | +/- | mapping strand of R1 (RNA-end), the actual cDNA strand is opposite |
| 7 | strand2 | +/- | mapping strand of R2 (DNA-end) |
| 8 | pair_type | string | pairtools defined pair_type |
| 9 | mapq1 | int | bwa MAPQ value of R1 (RNA-end) alignment |
| 10 | mapq2 | int | bwa MAPQ value of R2 (DNA-end) alignment |
| 11 | cigar1 | string | bwa cigar of R1 (RNA-end) alignment |
| 12 | cigar2 | string | bwa cigar of R2 (DNA-end) alignment |
| 13 | frag1_start | int | start position of assigned restriction fragment of R1 (RNA-end) |
| 14 | frag1_end | int | end position of assigned restriction fragment of R1 (RNA-end) |
| 15 | dist1_rsite | int | distance between 5' end mapping position of R1 and the nearest restriction digestion site position |
| 16 | frag2_start | int | start position of assigned restriction fragment of R2 (DNA-end) |
| 17 | frag2_end | int | end position of assigned restriction fragment of R2 (DNA-end) |
| 18 | dist2_rsite | int | distance between 5' end mapping position of R2 and the nearest restriction digestion site position |
The pipeline summary log file¶
When the pipeline finished, it will also generate a simple pipeline summary log file, named as pipelineStats_*.log. It
reports the sequencing mapping QC result at the first line. If it shows “Sequence mapping QC failed”, your data might
have critical problem for further analysis. The other lines show stats number of total read pairs,
non-dup unique mapping read pairs, final valid read pairs and so on.
Here we describe each line of the log file (TAB separated text file).
Line 1:
Sequence mapping QCIt’s the sequencing mapping QC result, which can be
passedorfailed). It’s concluded based on the value of line 2 and 3. Only when both values of line 2 and 3 are greater than threshold 0.5, it can be concluded aspassed.Line 2:
(#unique_mapped_pairs + #single_side_unique_mapped)/#total_read_pairsMapping rate of at least one side of read pairs uniquely mapped to genome. It can be calculated by dividing the sum of line 5 and line 6 by line 4.
Line 3:
#total_valid_interactions/#unique_mapped_pairsThe ratio of retained read pairs of unique mapped read pairs after de-duplication and filtering. It can be calculated by dividing line 8 by line 6.
Line 4:
total_read_pairsTotal number of read pairs in the FASTQ data.
Line 5:
single_side_unique_mappedNumber of single side unique mapped read pairs.
Line 6:
unique_mapped_pairsNumber of unique mapped read pairs (both two side unique mapped).
Line 7:
non_dup_unique_mapped_parisNumber of non-duplicated unique mapped read pairs.
Line 8:
total_valid_interactionsNumber of final output valid read pairs (RNA-DNA interactions).
Line 9:
inter_chrNumber of inter-chromosomal interacted read pairs (two ends mapped to different chromosomes).
Line 10:
intra_chrNumber of intra-chromosomal interacted read pairs (two ends mapped to the same chromosome).
Here is the output pipelineStats_test_sample.log file in our example.
Sequence mapping QC passed
(#unique_mapped_pairs + #single_side_unique_mapped)/#total_read_pairs 0.785859
#total_valid_interactions/#nondup_unique_mapped_pairs 0.768195
total_read_pairs 900000
single_side_unique_mapped 3342
unique_mapped_pairs 703931
nondup_unique_mapped_pairs 694706
total_valid_interactions 533670
inter_chr 244208
intra_chr 289462